
生物分子模拟是解析生命机制、赋能药物研发的核心技术,然而量子力学精度与计算可扩展性的长期矛盾,始终是该领域发展的关键瓶颈。近日,上海创智学院和浙江大学AIDD团队在国际顶级期刊《Nature Communications》发表重磅研究成果,提出全新等变神经网络 LiTEN 及其基础模型 LiTEN-FF,凭借原创的线性张量四边形注意力机制,首次在物理一致性、建模精度和计算效率间实现最优平衡,为生物分子建模与药物设计打造了新一代高性能物理计算工具。该研究论文题为 “A scalable and quantum-accurate foundation model for biomolecular force fields via linearly tensorized quadrangle attention”,上海创智学院博士生苏群为本文第一作者。

研究背景
准确的原子模拟对于理解疾病机制、推进药物发现和设计生物材料至关重要。传统方法主要分为两类:经典力场计算效率高,能进行大规模分子动力学模拟,但由于参数固定,难以准确模拟键的重排、过渡态能量和细微构象变化;而量子化学方法(如密度泛函理论)能精确描述电子结构,但高昂的计算成本限制了其在复杂生物大分子动态模拟中的应用。近年来,基于量子化学数据训练的机器学习势(MLIPs)为解决这一难题提供了新途径。尽管取得了显著进展,目前的生物分子建模仍面临多重挑战。首先,现有数据集碎片化且依赖不同精度的从头算方法,在势能面中引入了偏差,阻碍了模型的迁移学习。其次,主流数据集对关键生物分子子空间(如碳水化合物、极性残基等)的表征不足,限制了模型在真实生物系统中的泛化能力。此外,现存模型陷入了精度与效率的困境:高阶等变模型物理一致性好但推理效率极低;而建构于笛卡尔坐标系的低阶模型速度快,却难以准确预测局部多体效应。因此,迫切需要一种在物理一致性、建模精度和计算效率之间取得最佳平衡的新型计算框架。
方法概述
为打破精度与效率之间的壁垒,研究团队开发了 LiTEN 框架。其核心创新是张量四元注意力(TQA)机制。该机制摒弃了传统的显式高阶张量乘积和球谐函数展开,转而通过向量间的点乘和叉乘重新参数化高阶张量结构,紧凑地编码相互作用并保持等变性。这一设计大幅降低了计算图复杂度,成功突破了Clebsch–Gordan系数计算带来的性能瓶颈,以线性复杂度高效捕获复杂的分子空间扭转及多体耦合关系。在此架构基础上,研究团队采用两阶段层次化训练策略构建了 LiTEN-FF 基础模型:首先在包含1600万个类药物分子构象的 nablaDFT 大规模数据集上进行预训练,以全面捕捉药物样分子的电子结构特征;随后在精度更高、包含200万个生物有机分子的 SPICE 数据集上进行微调。这种训练策略使 LiTEN-FF 具备了跨复杂化学空间与溶剂化环境的强大泛化能力。
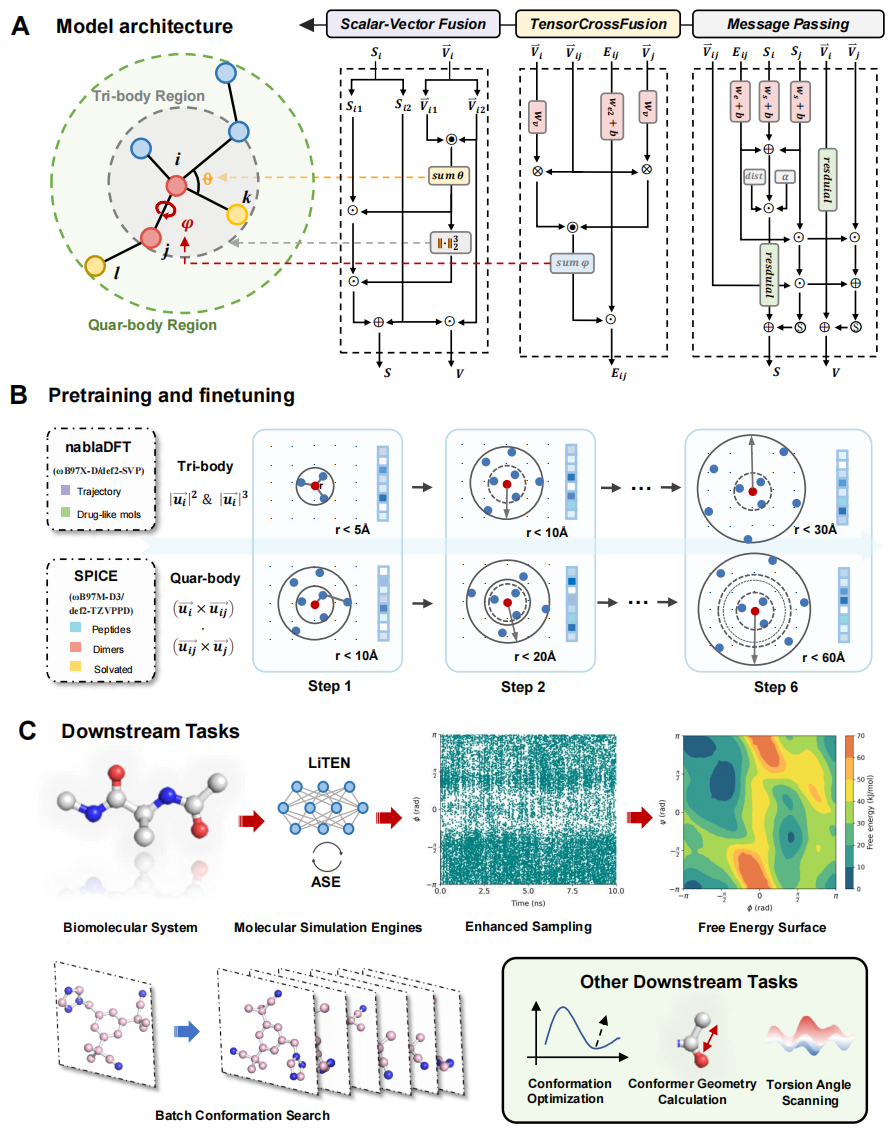
图1. LiTEN-FF方法概述
结果与讨论
基础测试集性能评估
研究团队在 rMD17、MD22 和 Chignolin 等标准基准测试集上系统评估了 LiTEN 的性能。结果显示,LiTEN 在绝大多数子集上达到了最先进(SOTA)水平,在能量和力的预测精度及训练推理速度上,全面超越了 MACE、NequIP 和 Allegro 等主流高精度方法。
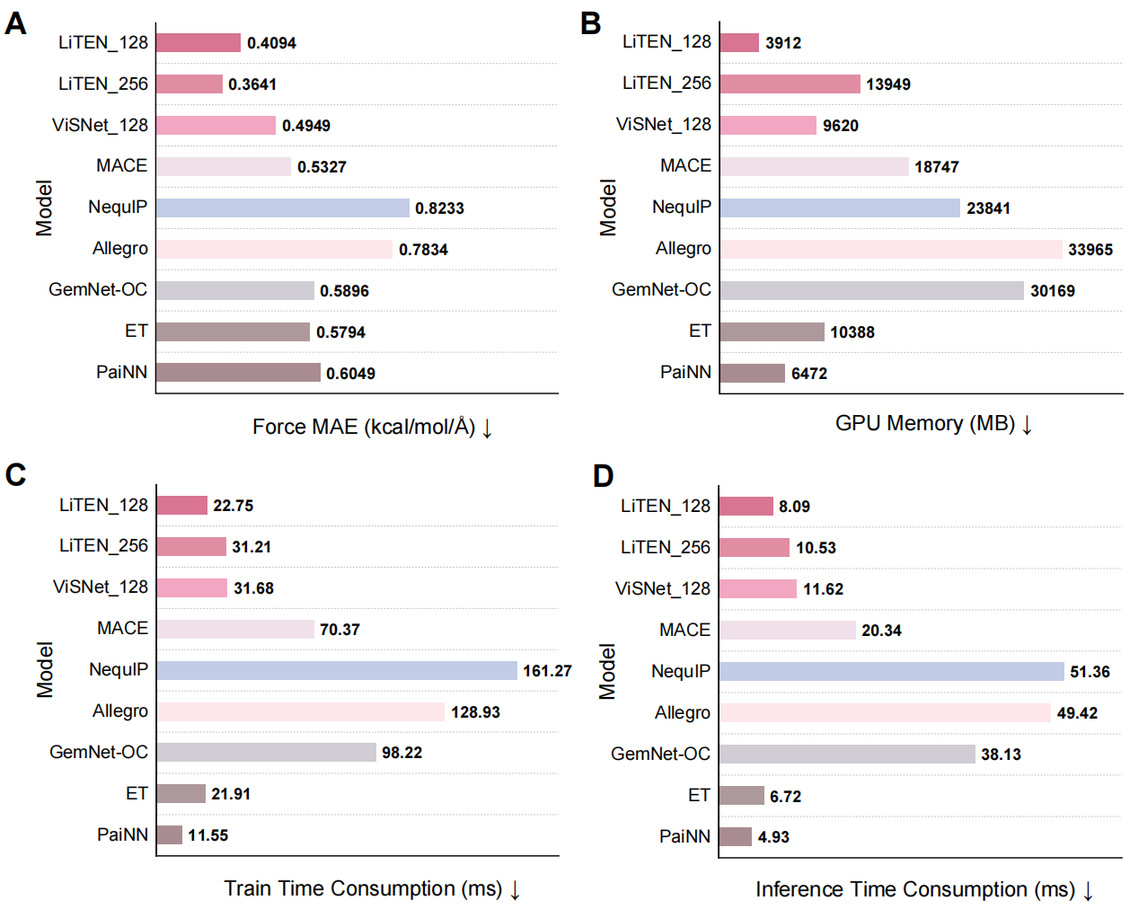
图2. 多种方法在Chignolin数据集上的综合评估
构象优化与动态几何建模能力
在真空体系的构象优化任务中,LiTEN-FF 达到了量子级精度,其优化出的分子结构与高精度 DFT 计算结果相比,平均均方根误差(RMSD)仅为 0.048 Å。在分子动力学模拟中,该模型能高保真地重现 DFT 级的键长和键角分布,KL 散度极低(0.001–0.01)。此外,在严苛的 TorsionNet206 扭转能基准测试中,LiTEN-FF 不仅优于现有的 MLIPs,还超越了中等级别 DFT 方法,相较于高精度 CCSD(T) 的平均绝对误差(MAE)仅为 0.19 kcal/mol,展现出精准刻画分子势能面的强大能力。
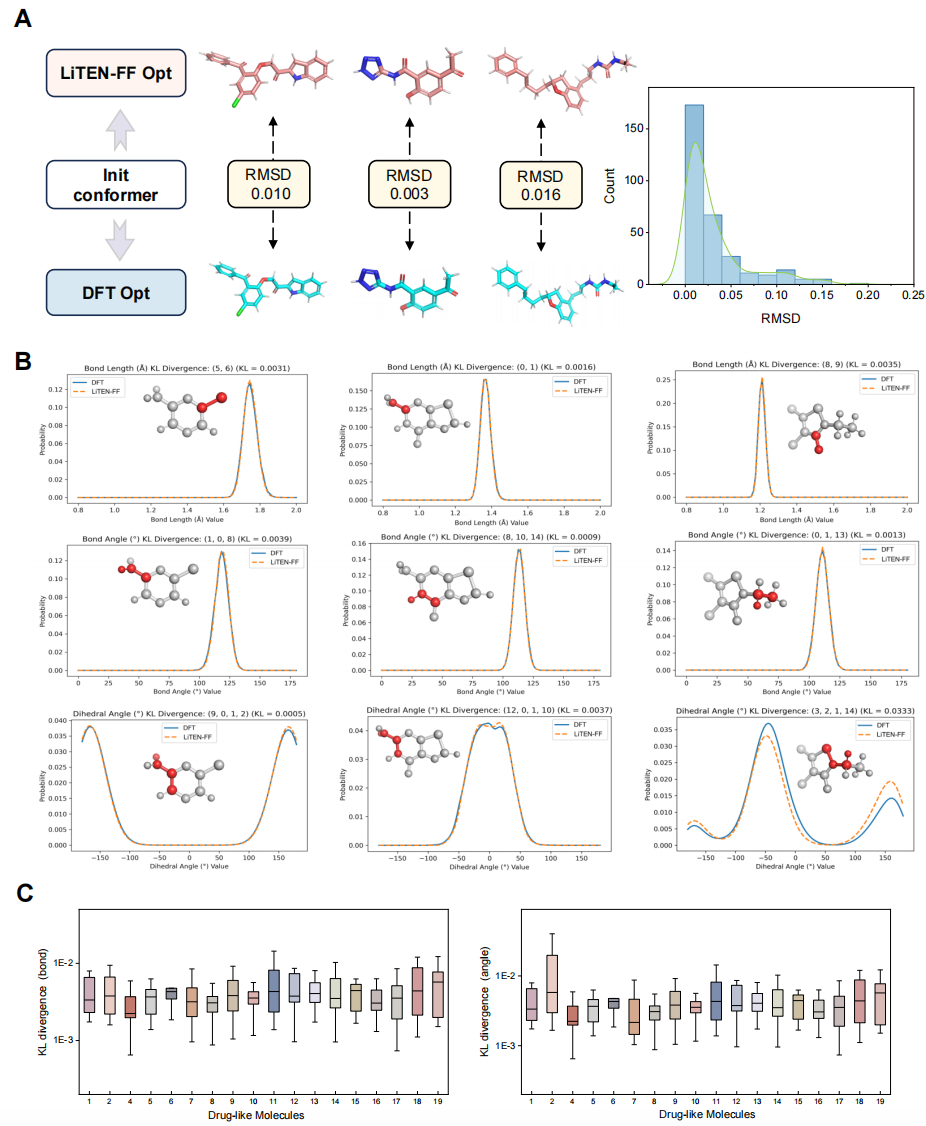
图3. LiTEN-FF 在真空体系中的应用与评估
大尺度体系的高效模拟与水溶液性质复现:
针对包含约1000个原子的大型分子体系,LiTEN-FF展现出卓越的计算可扩展性,其模拟速度比同类领先模型MACE-OFF快10倍以上。在确保极高推理速度的同时,该模型在周期性水盒子模拟中依然保持了严格的物理保真度,不仅准确复现了径向分布函数(RDF),还精确捕捉到了关键的水合层结构特征。

图4.周期性体系中的计算效率与性能
复杂液相体系的自由能景观重构
在更具挑战性的丙氨酸四肽(ala4)水溶液体系中,LiTEN-FF能够精准刻画分子的热力学动态行为。其生成的自由能表面(FES)景观与MACE-OFF高度一致,成功识别出四个关键的亚稳态构象盆地。此外,LiTEN-FF在捕获复杂构象异质性的同时,在大规模采样上展现出显著的效率优势,为真实溶剂环境下的生物大分子动力学模拟奠定了坚实基础。
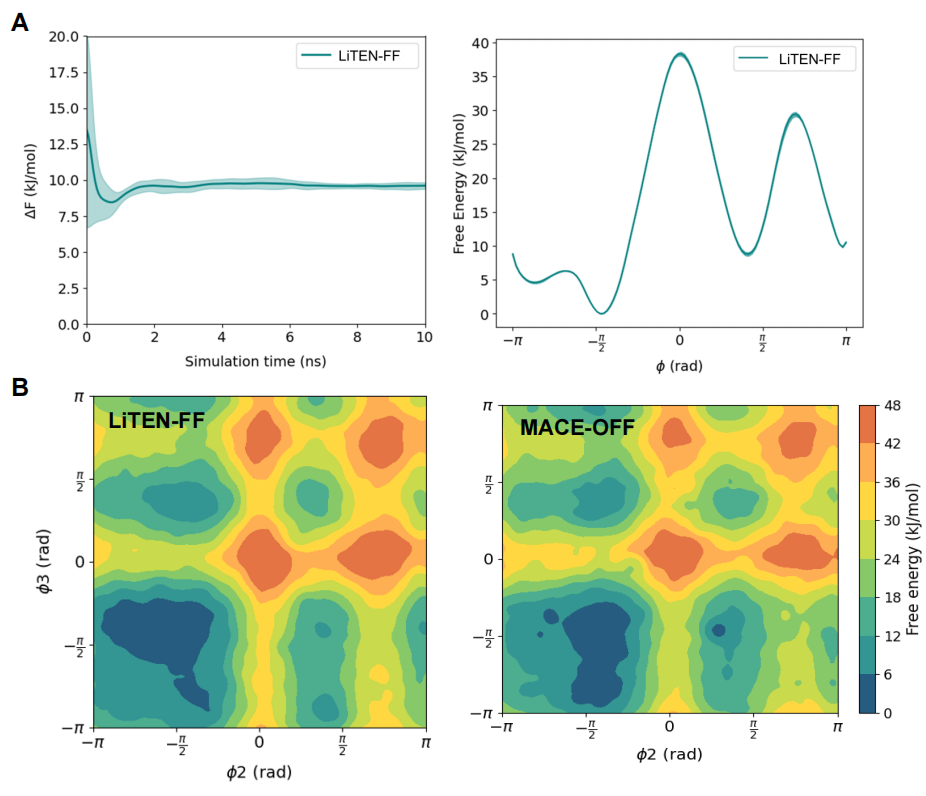
图5. ala4体系自由能景观的增强采样
批量构象搜索性能
通过部署退火和高温动力学模拟工作流,LiTEN-FF 能够快速探索并生成涵盖宽广能量盆地的多样化分子构象。凭借其独特的计算架构,当处理大量分子的构象搜索任务时,LiTEN-FF 展现出卓越的并行加速能力;随着分子批量的增加,生成速度可实现高达40倍的提升,极大推进了高通量药物筛选工作流的效率。
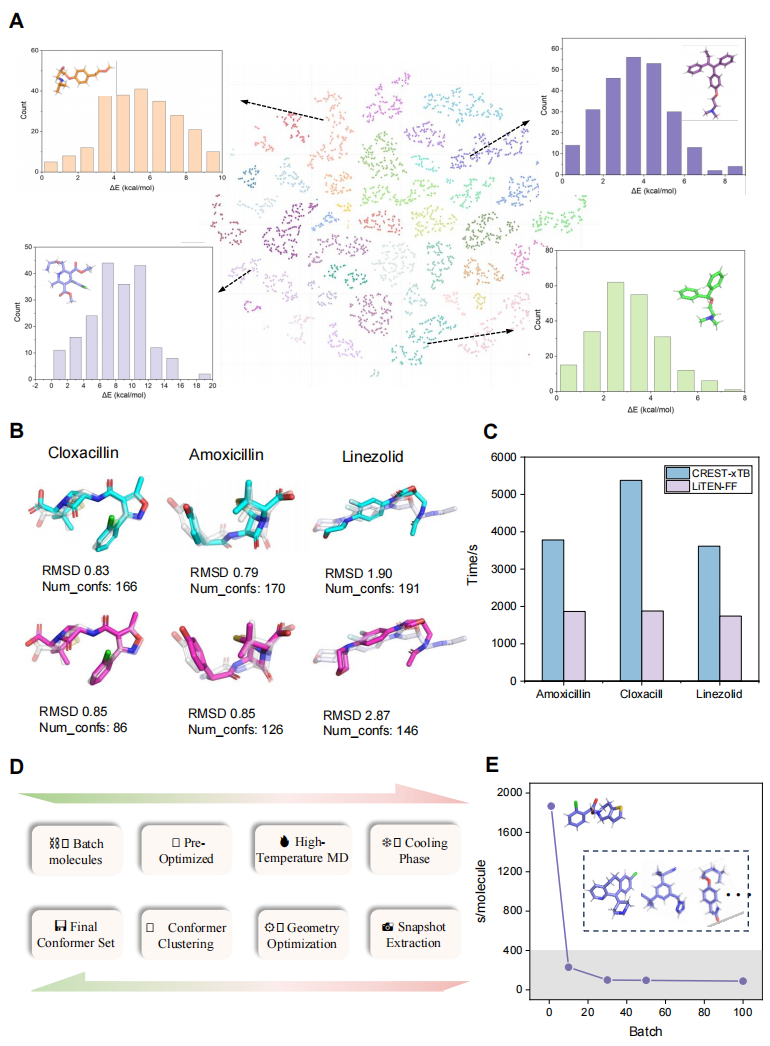
图6. 基于 LiTEN-FF 的构象搜索流程的可扩展性与效率
总结
总而言之,LiTEN-FF 的提出标志着分子模拟领域的重要进展。该框架将严格的物理一致性、精准的多体几何建模与高效的推理能力相结合,突破了机器学习力场在量子级精度与大规模计算效率之间长期存在的矛盾。作为面向生物分子体系的新一代基础力场模型,具备良好的跨化学空间泛化能力和溶剂环境适应性,在实际应用中,它可高效支持构象搜索、几何优化、自由能表面构建及复杂大分子动力学模拟,并在千原子规模体系中实现最高约十倍的计算加速。未来,随着其与蛋白质结构预测和分子生成模型的进一步融合,LiTEN-FF 有望成为 AI 分子模拟的重要基础引擎,在药物发现和材料科学等领域展现广阔应用前景。
原文链接
https://www.nature.com/articles/s41467-026-70377-4
